
Anti-HBA1 Antibody (CAB7322)
")
- SKU:
- CAB7322
- Product type:
- Antibody
- Reactivity:
- Human
- Mouse
- Rat
- Host Species:
- Rabbit
- Isotype:
- IgG
- Antibody Type:
- Polyclonal Antibody
- Research Area:
- Signal Transduction
")
Frequently bought together:
Description
| Antibody Name: | Anti-HBA1 Antibody |
| Antibody SKU: | CAB7322 |
| Antibody Size: | 20uL, 50uL, 100uL |
| Application: | WB IF |
| Reactivity: | Human, Mouse, Rat |
| Host Species: | Rabbit |
| Immunogen: | Recombinant fusion protein containing a sequence corresponding to amino acids 1-142 of human HBA1 (NP_000508.1). |
| Application: | WB IF |
| Recommended Dilution: | WB 1:500 - 1:2000 IF 1:50 - 1:100 |
| Reactivity: | Human, Mouse, Rat |
| Positive Samples: | MCF7, Mouse liver, Mouse blood cells, Rat liver, Rat blood cells |
| Immunogen: | Recombinant fusion protein containing a sequence corresponding to amino acids 1-142 of human HBA1 (NP_000508.1). |
| Purification Method: | Affinity purification |
| Storage Buffer: | Store at -20°C. Avoid freeze / thaw cycles. Buffer: PBS with 0.02% sodium azide, 50% glycerol, pH7.3. |
| Isotype: | IgG |
| Sequence: | MVLS PADK TNVK AAWG KVGA HAGE YGAE ALER MFLS FPTT KTYF PHFD LSHG SAQV KGHG KKVA DALT NAVA HVDD MPNA LSAL SDLH AHKL RVDP VNFK LLSH CLLV TLAA HLPA EFTP AVHA SLDK FLAS VSTV LTSK YR |
| Gene ID: | 3039 |
| Uniprot: | P69905 |
| Cellular Location: | |
| Calculated MW: | 15kDa |
| Observed MW: | 14kDa/25kDa |
| Synonyms: | HBA1, HBA-T3, HBH |
| Background: | The human alpha globin gene cluster located on chromosome 16 spans about 30 kb and includes seven loci: 5'- zeta - pseudozeta - mu - pseudoalpha-1 - alpha-2 - alpha-1 - theta - 3'. The alpha-2 (HBA2) and alpha-1 (HBA1) coding sequences are identical. These genes differ slightly over the 5' untranslated regions and the introns, but they differ significantly over the 3' untranslated regions. Two alpha chains plus two beta chains constitute HbA, which in normal adult life comprises about 97% of the total hemoglobin; alpha chains combine with delta chains to constitute HbA-2, which with HbF (fetal hemoglobin) makes up the remaining 3% of adult hemoglobin. Alpha thalassemias result from deletions of each of the alpha genes as well as deletions of both HBA2 and HBA1; some nondeletion alpha thalassemias have also been reported. |
| UniProt Protein Function: | HBA1: Involved in oxygen transport from the lung to the various peripheral tissues. Defects in HBA1 may be a cause of Heinz body anemias (HEIBAN). This is a form of non-spherocytic hemolytic anemia of Dacie type 1. After splenectomy, which has little benefit, basophilic inclusions called Heinz bodies are demonstrable in the erythrocytes. Before splenectomy, diffuse or punctate basophilia may be evident. Most of these cases are probably instances of hemoglobinopathy. The hemoglobin demonstrates heat lability. Heinz bodies are observed also with the Ivemark syndrome (asplenia with cardiovascular anomalies) and with glutathione peroxidase deficiency. Defects in HBA1 are the cause of alpha-thalassemia (A- THAL). The thalassemias are the most common monogenic diseases and occur mostly in Mediterranean and Southeast Asian populations. The hallmark of alpha-thalassemia is an imbalance in globin-chain production in the adult HbA molecule. The level of alpha chain production can range from none to very nearly normal levels. Deletion of both copies of each of the two alpha-globin genes causes alpha(0)-thalassemia, also known as homozygous alpha thalassemia. Due to the complete absence of alpha chains, the predominant fetal hemoglobin is a tetramer of gamma-chains (Bart hemoglobin) that has essentially no oxygen carrying capacity. This causes oxygen starvation in the fetal tissues leading to prenatal lethality or early neonatal death. The loss of three alpha genes results in high levels of a tetramer of four beta chains (hemoglobin H), causing a severe and life-threatening anemia known as hemoglobin H disease. Untreated, most patients die in childhood or early adolescence. The loss of two alpha genes results in mild alpha-thalassemia, also known as heterozygous alpha-thalassemia. Affected individuals have small red cells and a mild anemia (microcytosis). If three of the four alpha-globin genes are functional, individuals are completely asymptomatic. Some rare forms of alpha-thalassemia are due to point mutations (non- deletional alpha-thalassemia). The thalassemic phenotype is due to unstable globin alpha chains that are rapidly catabolized prior to formation of the alpha-beta heterotetramers. Alpha(0)-thalassemia is associated with non-immune hydrops fetalis, a generalized edema of the fetus with fluid accumulation in the body cavities due to non-immune causes. Non- immune hydrops fetalis is not a diagnosis in itself but a symptom, a feature of many genetic disorders, and the end-stage of a wide variety of disorders. Defects in HBA1 are the cause of hemoglobin H disease (HBH). HBH is a form of alpha-thalassemia due to the loss of three alpha genes. This results in high levels of a tetramer of four beta chains (hemoglobin H), causing a severe and life-threatening anemia. Untreated, most patients die in childhood or early adolescence. Belongs to the globin family. |
| UniProt Protein Details: | Protein type:Carrier Chromosomal Location of Human Ortholog: 16p13.3 Cellular Component: membrane; hemoglobin complex; extracellular region; cytosol Molecular Function:haptoglobin binding; protein binding; peroxidase activity; iron ion binding; heme binding; oxygen binding; oxygen transporter activity Biological Process: receptor-mediated endocytosis; response to hydrogen peroxide; hydrogen peroxide catabolic process; protein heterooligomerization; bicarbonate transport; oxygen transport Disease: Hemoglobin H Disease; Heinz Body Anemias; Alpha-thalassemia |
| NCBI Summary: | The human alpha globin gene cluster located on chromosome 16 spans about 30 kb and includes seven loci: 5'- zeta - pseudozeta - mu - pseudoalpha-1 - alpha-2 - alpha-1 - theta - 3'. The alpha-2 (HBA2) and alpha-1 (HBA1) coding sequences are identical. These genes differ slightly over the 5' untranslated regions and the introns, but they differ significantly over the 3' untranslated regions. Two alpha chains plus two beta chains constitute HbA, which in normal adult life comprises about 97% of the total hemoglobin; alpha chains combine with delta chains to constitute HbA-2, which with HbF (fetal hemoglobin) makes up the remaining 3% of adult hemoglobin. Alpha thalassemias result from deletions of each of the alpha genes as well as deletions of both HBA2 and HBA1; some nondeletion alpha thalassemias have also been reported. [provided by RefSeq, Jul 2008] |
| UniProt Code: | P69905 |
| NCBI GenInfo Identifier: | 57013850 |
| NCBI Gene ID: | 3040 |
| NCBI Accession: | P69905.2 |
| UniProt Secondary Accession: | P69905,P01922, Q1HDT5, Q3MIF5, Q53F97, Q96KF1, Q9NYR7 Q9UCM0, |
| UniProt Related Accession: | P69905 |
| Molecular Weight: | 15,258 Da |
| NCBI Full Name: | Hemoglobin subunit alpha |
| NCBI Synonym Full Names: | hemoglobin, alpha 2 |
| NCBI Official Symbol: | HBA2 |
| NCBI Official Synonym Symbols: | HBH; HBA-T2 |
| NCBI Protein Information: | hemoglobin subunit alpha; alpha globin; alpha-globin; alpha-2 globin; hemoglobin alpha chain |
| UniProt Protein Name: | Hemoglobin subunit alpha |
| UniProt Synonym Protein Names: | Alpha-globin; Hemoglobin alpha chain |
| Protein Family: | Hemoglobin |
| UniProt Gene Name: | HBA1 |
| UniProt Entry Name: | HBA_HUMAN |
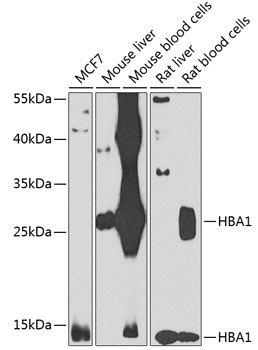 | Western blot analysis of extracts of various cell lines, using HBA1 antibody (CAB7322) at 1:1000 dilution._Secondary antibody: HRP Goat Anti-Rabbit IgG (H+L) (CABS014) at 1:10000 dilution._Lysates/proteins: 25ug per lane._Blocking buffer: 3% nonfat dry milk in TBST._Detection: ECL Enhanced Kit (RM00021)._Exposure time: 30s. |
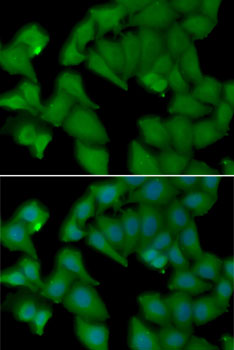 | Immunofluorescence analysis of MCF7 cells using HBA1 antibody (CAB7322). Blue: DAPI for nuclear staining. |
Related Products
")
Anti-HBA1 Antibody (CAB14551)
")

Human HBA1 / Hemoglobin subunit alpha ELISA Kit
 ELISA Kit")
Human Hemoglobin subunit alpha (HBA1) ELISA Kit
")
 ELISA Kit")
Human anti-EPO antibody(anti-Erythropoietin antibody) ELISA Kit
