
Anti-CPT2 Antibody (CAB0653)
")
- SKU:
- CAB0653
- Product type:
- Antibody
- Reactivity:
- Human
- Mouse
- Rat
- Host Species:
- Rabbit
- Isotype:
- IgG
- Antibody Type:
- Monoclonal Antibody
- Research Area:
- Metabolism
")
Frequently bought together:
Description
| Antibody Name: | Anti-CPT2 Antibody |
| Antibody SKU: | CAB0653 |
| Antibody Size: | 20uL, 50uL, 100uL |
| Application: | WB |
| Reactivity: | Human, Mouse, Rat |
| Host Species: | Rabbit |
| Immunogen: | A synthesized peptide derived from human CPT2 |
| Application: | WB |
| Recommended Dilution: | WB 1:500 - 1:2000 |
| Reactivity: | Human, Mouse, Rat |
| Positive Samples: | HeLa, 293T, HepG2, Mouse liver, Mouse heart, Rat liver, Rat heart |
| Immunogen: | A synthesized peptide derived from human CPT2 |
| Purification Method: | Affinity purification |
| Storage Buffer: | Store at -20°C. Avoid freeze / thaw cycles. Buffer: PBS with 0.02% sodium azide, 0.05% BSA, 50% glycerol, pH7.3. |
| Isotype: | IgG |
| Sequence: | Email for sequence |
| Gene ID: | 1376 |
| Uniprot: | P23786 |
| Cellular Location: | |
| Calculated MW: | 74kDa |
| Observed MW: | 70KDa |
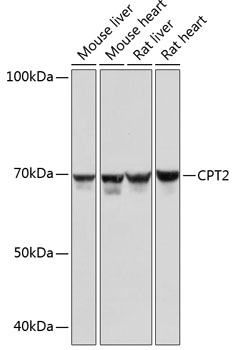 | Western blot - CPT2 Rabbit mAb (CAB0653) |
| Synonyms: | CPT1, CPTASE, IIAE4 |
| Background: | The protein encoded by this gene is a nuclear protein which is transported to the mitochondrial inner membrane. Together with carnitine palmitoyltransferase I, the encoded protein oxidizes long-chain fatty acids in the mitochondria. Defects in this gene are associated with mitochondrial long-chain fatty-acid (LCFA) oxidation disorders. [provided by RefSeq, Jul 2008] |
| UniProt Protein Function: | CPT2: Defects in CPT2 are the cause of carnitine palmitoyltransferase 2 deficiency late-onset (CPT2D); also known as CPT-II deficiency or CPT2 deficiency. CPT2D is an autosomal recessive disorder characterized by recurrent myoglobinuria, episodes of muscle pain, stiffness, and rhabdomyolysis. These symptoms are triggered by prolonged exercise, fasting or viral infection and patients are usually young adults. In addition to this classical, late-onset, muscular type, a hepatic or hepatocardiomuscular form has been reported in infants. Clinical pictures in these children or neonates include hypoketotic hypoglycemia, liver dysfunction, cardiomyopathy and sudden death. Defects in CPT2 are the cause of carnitine palmitoyltransferase 2 deficiency infantile (CPT2DI). A disorder of mitochondrial long-chain fatty acid oxidation characterized by hepatic or hepato-cardio-muscular manifestations with onset in infancy. Clinical features include hypoketotic hypoglycemia, lethargy, seizures, hepatomegaly, liver dysfunction, cardiomegaly and dilated cardiomyopathy. Defects in CPT2 are the cause of carnitine palmitoyltransferase 2 deficiency lethal neonatal (CPT2D-LN); also known as lethal neonatal CPT-II deficiency. It is a lethal neonatal form of CPT2D. This rarely presentation is antenatal with cerebral periventricular cysts and cystic dysplastic kidneys. The clinical variability of the disease is likely attributed to the variable residual enzymatic activity. Defects in CPT2 are a cause of susceptibility to encephalopathy acute infection-induced type 4 (IIAE4). A severe neurologic complication of an infection. It manifests within days in otherwise healthy children after common viral infections, without evidence of viral infection of the brain or inflammatory cell infiltration. In affected children, high- grade fever is accompanied within 12 to 48 hours by febrile convulsions, often leading to coma, multiple-organ failure, brain edema, and high morbidity and mortality. The infections are usually viral, particularly influenza, although other viruses and even mycoplasma have been found to cause the disorder. Polymorphic variants in CPT2 can confer susceptibility to infection-induced encepalopathy. These variants do not cause classical carnitine palmitoyltransferase 2 deficiency, and patients harboring any of them are asymptomatic most of the time. However, they are prone to viral infection (high fever)-related encephalopathy (PubMed:21697855). Belongs to the carnitine/choline acetyltransferase family. |
| UniProt Protein Details: | Protein type:EC 2.3.1.21; Mitochondrial; Transferase; Lipid Metabolism - fatty acid Chromosomal Location of Human Ortholog: 1p32 Cellular Component: nucleoplasm; mitochondrion; mitochondrial inner membrane; nucleolus Molecular Function:carnitine O-palmitoyltransferase activity Biological Process: fatty acid beta-oxidation; cellular lipid metabolic process; carnitine shuttle Disease: Carnitine Palmitoyltransferase Ii Deficiency, Lethal Neonatal; Carnitine Palmitoyltransferase Ii Deficiency, Infantile; Encephalopathy, Acute, Infection-induced, Susceptibility To, 4; Carnitine Palmitoyltransferase Ii Deficiency, Late-onset |
| NCBI Summary: | The protein encoded by this gene is a nuclear protein which is transported to the mitochondrial inner membrane. Together with carnitine palmitoyltransferase I, the encoded protein oxidizes long-chain fatty acids in the mitochondria. Defects in this gene are associated with mitochondrial long-chain fatty-acid (LCFA) oxidation disorders. [provided by RefSeq, Jul 2008] |
| UniProt Code: | P23786 |
| NCBI GenInfo Identifier: | 416836 |
| NCBI Gene ID: | 1376 |
| NCBI Accession: | P23786.2 |
| UniProt Related Accession: | P23786 |
| Molecular Weight: | |
| NCBI Full Name: | Carnitine O-palmitoyltransferase 2, mitochondrial |
| NCBI Synonym Full Names: | carnitine palmitoyltransferase 2 |
| NCBI Official Symbol: | CPT2 |
| NCBI Official Synonym Symbols: | CPT1; IIAE4; CPTASE |
| NCBI Protein Information: | carnitine O-palmitoyltransferase 2, mitochondrial |
| UniProt Protein Name: | Carnitine O-palmitoyltransferase 2, mitochondrial |
| UniProt Synonym Protein Names: | Carnitine palmitoyltransferase II; CPT II |
| Protein Family: | Splenin |
| UniProt Gene Name: | CPT2 |
| UniProt Entry Name: | CPT2_HUMAN |
Related Products
")
Anti-CPT2 Antibody (CAB12426)
")
 ELISA Kit")
Human anti-EPO antibody(anti-Erythropoietin antibody) ELISA Kit

")
 ELISA Kit")