
Gba Antibody (PACO48914)
")
- SKU:
- PACO48914
- Product type:
- Antibody
- Reactivity:
- Mouse
- Host Species:
- Rabbit
- Isotype:
- IgG
- Application:
- ELISA
- Application:
- WB
- Antibody type:
- Polyclonal
- Conjugation:
- Unconjugated
")
Description
| Antibody Name: | Gba Antibody (PACO48914) |
| Antibody SKU: | PACO48914 |
| Size: | 50ug |
| Host Species: | Rabbit |
| Tested Applications: | ELISA, WB |
| Recommended Dilutions: | ELISA:1:2000-1:10000, WB:1:500-1:5000 |
| Species Reactivity: | Mouse |
| Immunogen: | Recombinant Mouse Glucosylceramidase protein (20-515AA) |
| Form: | Liquid |
| Storage Buffer: | Preservative: 0.03% Proclin 300 Constituents: 50% Glycerol, 0.01M PBS, PH 7.4 |
| Purification Method: | >95%, Protein G purified |
| Clonality: | Polyclonal |
| Isotype: | IgG |
| Conjugate: | Non-conjugated |
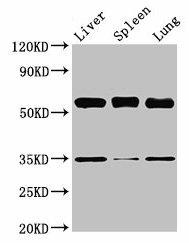 | Western Blot. Positive WB detected in: Mouse liver tissue, Mouse spleen tissue, Mouse lung tissue. All lanes: Gba antibody at 3.5µg/ml. Secondary. Goat polyclonal to rabbit IgG at 1/50000 dilution. Predicted band size: 58 kDa. Observed band size: 58 kDa.. |
| Background: | extracellular exosome, extracellular space, lysosomal lumen, lysosomal membrane, glucosylceramidase activity, hydrolase activity, receptor binding, cellular response to starvation, cellular response to tumor necrosis factor, ceramide biosynthetic process |
| Synonyms: | Glucosylceramidase (EC 3.2.1.45) (acid, beta-glucosidase) (Beta-glucocerebrosidase) (D-glucosyl-N-acylsphingosine glucohydrolase), Gba |
| UniProt Protein Function: | GBA: Defects in GBA are the cause of Gaucher disease (GD); also known as glucocerebrosidase deficiency. GD is the most prevalent lysosomal storage disease, characterized by accumulation of glucosylceramide in the reticulo-endothelial system. Different clinical forms are recognized depending on the presence (neuronopathic forms) or absence of central nervous system involvement, severity and age of onset. Defects in GBA are the cause of Gaucher disease type 1 (GD1); also known as adult non-neuronopathic Gaucher disease. GD1 is characterized by hepatosplenomegaly with consequent anemia and thrombopenia, and bone involvement. The central nervous system is not involved. Defects in GBA are the cause of Gaucher disease type 2 (GD2); also known as acute neuronopathic Gaucher disease. GD2 is the most severe form and is universally progressive and fatal. It manifests soon after birth, with death generally occurring before patients reach two years of age. Defects in GBA are the cause of Gaucher disease type 3 (GD3); also known as subacute neuronopathic Gaucher disease. GD3 has central nervous manifestations. Defects in GBA are the cause of Gaucher disease type 3C (GD3C); also known as pseudo-Gaucher disease or Gaucher-like disease. Defects in GBA are the cause of Gaucher disease perinatal lethal (GDPL). It is a distinct form of Gaucher disease type 2, characterized by fetal onset. Hydrops fetalis, in utero fetal death and neonatal distress are prominent features. When hydrops is absent, neurologic involvement begins in the first week and leads to death within 3 months. Hepatosplenomegaly is a major sign, and is associated with ichthyosis, arthrogryposis, and facial dysmorphism. Perinatal lethal Gaucher disease is associated with non-immune hydrops fetalis, a generalized edema of the fetus with fluid accumulation in the body cavities due to non-immune causes. Non-immune hydrops fetalis is not a diagnosis in itself but a symptom, a feature of many genetic disorders, and the end-stage of a wide variety of disorders. Defects in GBA contribute to susceptibility to Parkinson disease (PARK). A complex neurodegenerative disorder characterized by bradykinesia, resting tremor, muscular rigidity and postural instability. Additional features are characteristic postural abnormalities, dysautonomia, dystonic cramps, and dementia. The pathology of Parkinson disease involves the loss of dopaminergic neurons in the substantia nigra and the presence of Lewy bodies (intraneuronal accumulations of aggregated proteins), in surviving neurons in various areas of the brain. The disease is progressive and usually manifests after the age of 50 years, although early-onset cases (before 50 years) are known. The majority of the cases are sporadic suggesting a multifactorial etiology based on environmental and genetic factors. However, some patients present with a positive family history for the disease. Familial forms of the disease usually begin at earlier ages and are associated with atypical clinical features. Belongs to the glycosyl hydrolase 30 family. 3 isoforms of the human protein are produced by alternative splicing. |
| UniProt Protein Details: | Protein type:EC 3.2.1.45; Glycan Metabolism - other glycan degradation; Hydrolase; Lipid Metabolism - sphingolipid Cellular Component: extracellular space; lysosomal lumen; lysosomal membrane Molecular Function:glucosylceramidase activity; hydrolase activity; receptor binding Biological Process: cellular response to starvation; ceramide biosynthetic process; glucosylceramide catabolic process; negative regulation of cellular protein metabolic process; negative regulation of interleukin-6 production; negative regulation of MAP kinase activity; negative regulation of protein homooligomerization; positive regulation of proteasomal ubiquitin-dependent protein catabolic process; positive regulation of protein amino acid dephosphorylation; positive regulation of protein complex disassembly; positive regulation of protein metabolic process; regulation of cellular protein metabolic process; regulation of protein metabolic process; regulation of water loss via skin; response to estrogen stimulus; response to glucocorticoid stimulus; response to pH; response to testosterone stimulus; skin morphogenesis; sphingosine biosynthetic process |
| UniProt Code: | P17439 |
| NCBI GenInfo Identifier: | 116734815 |
| NCBI Gene ID: | 14466 |
| NCBI Accession: | NP_001070879.1 |
| UniProt Secondary Accession: | P17439,Q78NR7, |
| UniProt Related Accession: | P17439 |
| Molecular Weight: | |
| NCBI Full Name: | glucosylceramidase |
| NCBI Synonym Full Names: | glucosidase, beta, acid |
| NCBI Official Symbol: | Gba |
| NCBI Official Synonym Symbols: | GC; GBA1; GLUC; GCase; betaGC |
| NCBI Protein Information: | glucosylceramidase |
| UniProt Protein Name: | Glucosylceramidase |
| UniProt Synonym Protein Names: | Acid beta-glucosidase; Beta-glucocerebrosidase; D-glucosyl-N-acylsphingosine glucohydrolase |
| Protein Family: | Glucosylceramidase |
| UniProt Gene Name: | Gba |
| UniProt Entry Name: | GLCM_MOUSE |
Related Products
")
Anti-GBAS Antibody (CAB17888)
")
")
 ELISA Kit")

Mouse Glucosylceramidase / GBA ELISA Kit
 ELISA Kit")