
Anti-HTT Antibody (CAB16872)
")
- SKU:
- CAB16872
- Product type:
- Antibody
- Reactivity:
- Human
- Mouse
- Rat
- Host Species:
- Rabbit
- Isotype:
- IgG
- Antibody Type:
- Polyclonal Antibody
- Research Area:
- Cell Death
")
Description
| Antibody Name: | Anti-HTT Antibody |
| Antibody SKU: | CAB16872 |
| Antibody Size: | 20uL, 50uL, 100uL |
| Application: | WB IHC IF |
| Reactivity: | Human, Mouse, Rat |
| Host Species: | Rabbit |
| Immunogen: | A synthetic peptide of human HTT. |
| Application: | WB IHC IF |
| Recommended Dilution: | WB 1:500 - 1:2000 IHC 1:50 - 1:200 IF 1:50 - 1:200 |
| Reactivity: | Human, Mouse, Rat |
| Positive Samples: | HeLa, SH-SY5Y, U-251MG, Mouse brain |
| Immunogen: | A synthetic peptide of human HTT. |
| Purification Method: | Affinity purification |
| Storage Buffer: | Store at -20°C. Avoid freeze / thaw cycles. Buffer: PBS with 0.02% sodium azide, 50% glycerol, pH7.3. |
| Isotype: | IgG |
| Sequence: | Email for sequence |
| Gene ID: | 3064 |
| Uniprot: | P42858 |
| Cellular Location: | Cytoplasm, Nucleus |
| Calculated MW: | 347kDa |
| Observed MW: | 347kDa |
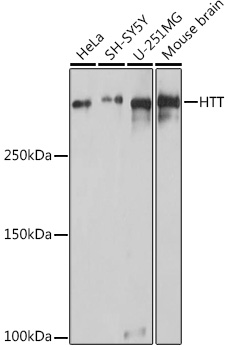 | Western blot - HTT Rabbit pAb (CAB16872) |
| Synonyms: | HD, IT15, HTT, Huntingtin |
| Background: | Huntingtin is a disease gene linked to Huntington's disease, a neurodegenerative disorder characterized by loss of striatal neurons. This is thought to be caused by an expanded, unstable trinucleotide repeat in the huntingtin gene, which translates as a polyglutamine repeat in the protein product. A fairly broad range of trinucleotide repeats (9-35) has been identified in normal controls, and repeat numbers in excess of 40 have been described as pathological. The huntingtin locus is large, spanning 180 kb and consisting of 67 exons. The huntingtin gene is widely expressed and is required for normal development. It is expressed as 2 alternatively polyadenylated forms displaying different relative abundance in various fetal and adult tissues. The larger transcript is approximately 13.7 kb and is expressed predominantly in adult and fetal brain whereas the smaller transcript of approximately 10.3 kb is more widely expressed. The genetic defect leading to Huntington's disease may not necessarily eliminate transcription, but may confer a new property on the mRNA or alter the function of the protein. One candidate is the huntingtin-associated protein-1, highly expressed in brain, which has increased affinity for huntingtin protein with expanded polyglutamine repeats. This gene contains an upstream open reading frame in the 5' UTR that inhibits expression of the huntingtin gene product through translational repression. |
| UniProt Protein Function: | Huntingtin: may play a role in microtubule-mediated transport or vesicle function. Widely expressed with the highest level of expression in the brain (nerve fibers, varicosities, and nerve endings). In the brain, the regions where it can be mainly found are the cerebellar cortex, the neocortex, the striatum, and the hippocampal formation. Defects are the cause of Huntington's disease, a neurodegenerative disorder characterized by loss of striatal neurons. This is thought to be caused by an expanded, unstable trinucleotide repeat in the Huntingtin gene, which translates as a polyglutamine repeat in the protein product. The Huntingtin locus is large, spanning 180 kb and consisting of 67 exons. The Huntingtin gene is widely expressed and is required for normal development. It is expressed as 2 alternatively polyadenylated isoforms displaying different relative abundance in various fetal and adult tissues. |
| UniProt Protein Details: | Protein type:Cytoskeletal Chromosomal Location of Human Ortholog: 4p16.3 Cellular Component: nucleoplasm; Golgi apparatus; protein complex; cytoplasmic vesicle membrane; mitochondrion; axon; endoplasmic reticulum; late endosome; dendrite; cytoplasm; autophagic vacuole; inclusion body; cytosol; nucleus Molecular Function:identical protein binding; protein binding; p53 binding; dynein intermediate chain binding; beta-tubulin binding; diazepam binding; transcription factor binding Biological Process: ER to Golgi vesicle-mediated transport; citrulline metabolic process; paraxial mesoderm formation; regulation of protein phosphatase type 2A activity; regulation of synaptic plasticity; locomotory behavior; determination of adult life span; endosome transport; anterior/posterior pattern formation; L-glutamate import; regulation of mitochondrial membrane potential; establishment of mitotic spindle orientation; protein import into nucleus; organ development; quinolinate biosynthetic process; retrograde vesicle-mediated transport, Golgi to ER; vesicle transport along microtubule; visual learning; negative regulation of neuron apoptosis; Golgi organization and biogenesis; grooming behavior; endoplasmic reticulum organization and biogenesis; positive regulation of inositol-1,4,5-triphosphate receptor activity; striatum development; axon cargo transport; cell aging; olfactory lobe development; social behavior; lactate biosynthetic process from pyruvate; neuron apoptosis; iron ion homeostasis; insulin secretion; dopamine receptor signaling pathway; hormone metabolic process; neuron development; spermatogenesis; regulation of mitochondrial membrane permeability; response to calcium ion; neural plate formation; urea cycle Disease: Huntington Disease |
| NCBI Summary: | Huntingtin is a disease gene linked to Huntington's disease, a neurodegenerative disorder characterized by loss of striatal neurons. This is thought to be caused by an expanded, unstable trinucleotide repeat in the huntingtin gene, which translates as a polyglutamine repeat in the protein product. A fairly broad range in the number of trinucleotide repeats has been identified in normal controls, and repeat numbers in excess of 40 have been described as pathological. The huntingtin locus is large, spanning 180 kb and consisting of 67 exons. The huntingtin gene is widely expressed and is required for normal development. It is expressed as 2 alternatively polyadenylated forms displaying different relative abundance in various fetal and adult tissues. The larger transcript is approximately 13.7 kb and is expressed predominantly in adult and fetal brain whereas the smaller transcript of approximately 10.3 kb is more widely expressed. The genetic defect leading to Huntington's disease may not necessarily eliminate transcription, but may confer a new property on the mRNA or alter the function of the protein. One candidate is the huntingtin-associated protein-1, highly expressed in brain, which has increased affinity for huntingtin protein with expanded polyglutamine repeats. This gene contains an upstream open reading frame in the 5' UTR that inhibits expression of the huntingtin gene product through translational repression. [provided by RefSeq, Jul 2008] |
| UniProt Code: | P42858 |
| NCBI GenInfo Identifier: | 296434520 |
| NCBI Gene ID: | 3064 |
| NCBI Accession: | P42858.2 |
| UniProt Secondary Accession: | P42858,Q9UQB7, |
| UniProt Related Accession: | P42858 |
| Molecular Weight: | |
| NCBI Full Name: | Huntingtin |
| NCBI Synonym Full Names: | huntingtin |
| NCBI Official Symbol: | HTT |
| NCBI Official Synonym Symbols: | HD; IT15 |
| NCBI Protein Information: | huntingtin; huntington disease protein |
| UniProt Protein Name: | Huntingtin |
| UniProt Synonym Protein Names: | Huntington disease protein; HD protein |
| Protein Family: | HD protein |
| UniProt Gene Name: | HTT |
| UniProt Entry Name: | HD_HUMAN |
Related Products
")
 ELISA Kit")
Human anti-EPO antibody(anti-Erythropoietin antibody) ELISA Kit

 ELISA Kit")
Human Anti-DSG3 antibody(anti-Desmoglein-3 antibody) ELISA Kit
")